Chapter 2: DFTB+ Calculations¶
Here we illustrate some standard procedures that can be performed by the DFTB+ program in conjunction with DJMol platform. At present there are three distinct file types can be opened for DFTB+ calculations, viz., [1] dftb_in.hsd, <.xyz> and <.gen> files. The first one is the default unique file type for the program and the last two file extensions can be converted into <dftb_in.hsd> file by using a script writer (see below section).
DFTB+ Script Writer¶
If one opens a <.xyz> or <.hsd> file, for visualizing the structure, subsequently one can also try to make a corresponding <dftb_in.hsd> file using a DFTB+ script writer program. Using this, a user can make a basic <dftb_in.hsd> script for a range of jobs, including, geometry optimization, calculating excited states or Hessian etc. It can be retrieved by:
Set Up ➤ DFTB Scripting
And the generated <dftb_in.hsd> file is kept in <Input> folder. Note that the atomic information including its geometry is directly taken from <gen> data of the currently loaded file.
Note that, the Script-Writer module will write <TEMPdftb_in.hsd> into the <Input> folder. It is strongly recommended that, a user should always try to inspect/modify this script before its submission, if it is necessary.
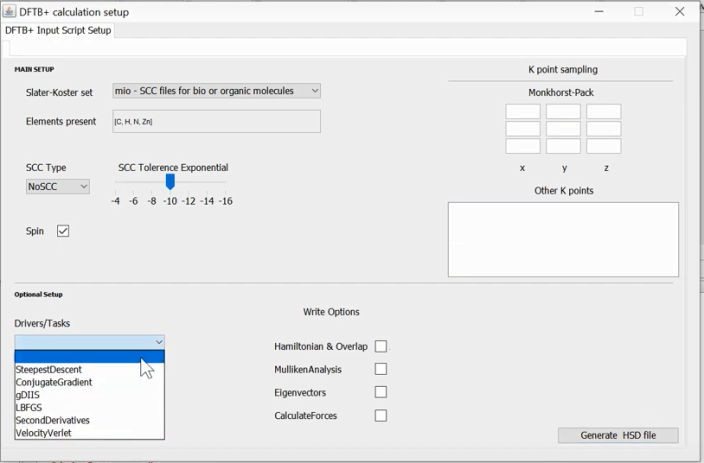
A screen shot of the script writer tool for the DFTB+ program.
Executing DFTB+ Scripts¶
Running DFTB+ calculator is possible when you load a <dftb_in.hsd> file. Make sure that the <Input> folder should have this file before the execution and this file can be executed as:
Execute ➤ Run DFTB+
And if necessary, this Run can be killed by
Execute ➤ Stop DFTB+
The generated/loaded files are usually placed in the <DFTB_Scratch> folder. And these files are also sent back as a Zip file, to the directory where your current <hsd> file resides.
DFTB+ generated data can be analyzed or visualized by the program. Main data analyzers are given below.
Force Components¶
DFTB+ out file contains information about Cartesian force components (in atomic units) and this can be visualized as a line chart as shown below. The <detailed.out> file can be opened by:
Tools ➤ ForceComponents
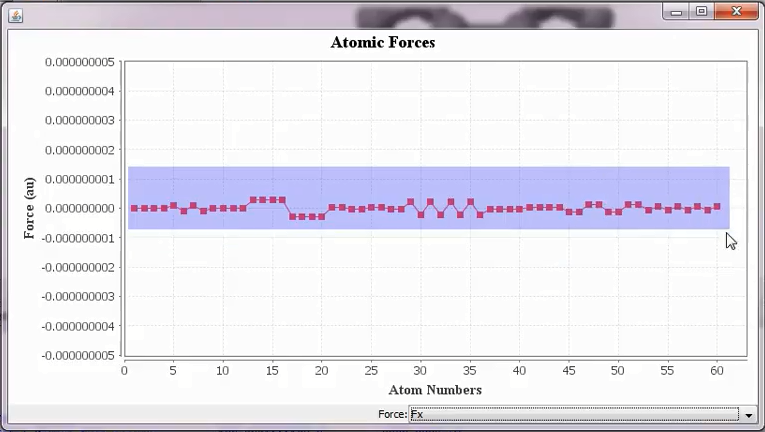
The x components of the Cartesian forces of the Fullerene molecule after the geometry optimization. The shaded portion can be easily zoomed in for more details.¶
Molecular Orbitals and Energy Levels¶
MO information (including density and density difference files) are stored in the .CUBE format. In order to get cube files one needs to ensure that dftb_in.hsd file contains relevant keywords (WriteDetailedXML and WriteEigenvectors, see example directory for a template file). After running this file it will produce 3 out files (detailed.xml, charges.bin, and eigenvec.bin) and these files should be transferred from <DFTB_Scratch> directory to <ModesBinary> folder. Then invoke:
Execute ➤ Run Waveplot
From this window, select the proper SK file set (look at the combo box entitled, Specify SK set and Generate HSD file) and press on the button <Generate Waveplot File>. This will produce the needed HSD file for the Waveplot.exe binary file. View this file from the second tab and edit it if needed. Then invoke the <Calculate> button to produce the necessary CUBE file. Note that it might take some minutes to complete the task (depending on the size of the system).
To visualize the cubes file, first copy the Full Path of the folder, <ModesBinary> and then go to:
Tools ➤ View Cube
Then click on the button, <Open Directory> and insert the recently copied Full Path into it and select a CUBE file. This will list all the CUBE files into the table, and this can be visualized easily by selecting the needed file name from the table and by clicking on the <Open> button. Optionally one can also use JMol scripting facility for more advanced file visualizations or manipulations.
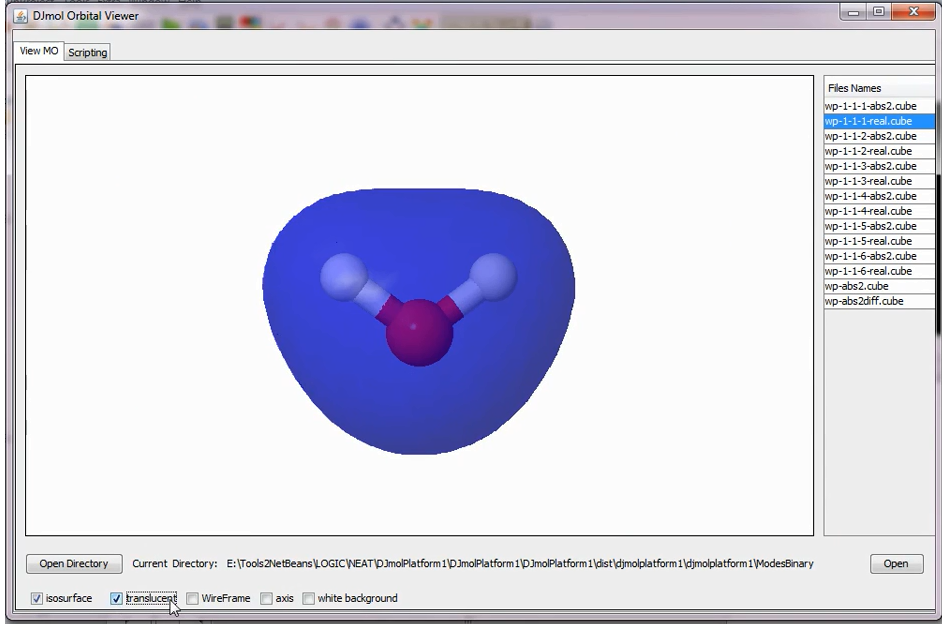
Visual of one of the MOs from the list of CUBE files.¶
To view only MO energy levels and its degeneracies, one can use the <detailed.out> file and this can be called by:
Tools ➤ MO Levels
The green lines represent occupied and red lines represent the un occupied levels. For accurate calculations (which use tight SCC parameters) one can also look at the degeneracies levels.
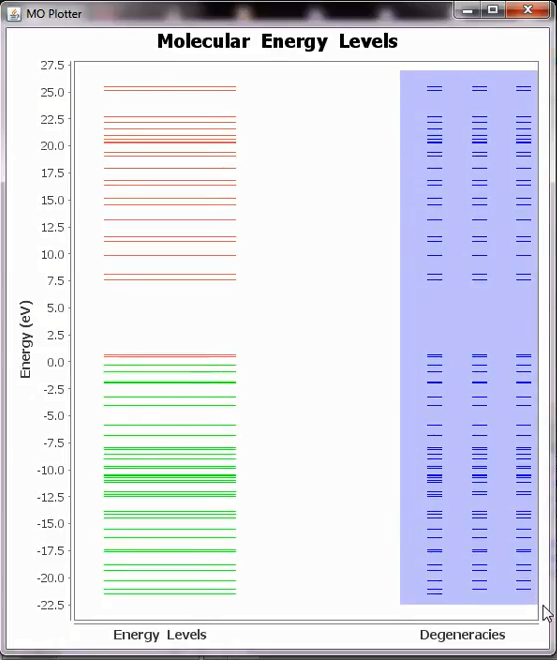
MO energy levels from the detailed.out file.¶
UV-Visible Spectrum Convolution¶
DFTB+ uses TD-DFTB for calculating oscillator strength of the optical excitations and this is stored in SPX.dat file. An add-on is used for reading this data and this can be invoked by:
Tools ➤ UV-Vis Spectrum
Select a SPX.dat file to proceed. This will plot the oscillator strengths (vertical red lines) and its fitted function ( broadened through convolution with a Gaussian, Lorentzian function or its linear combination, known as pseudo-Voigt functions - shown by blue curves). By default it uses eV however one can also obtain an equivalent spectrum using nm units (this creates a separate plot). Note that the oscillator strength is dimensionless quantity and it is described in the y axis.
By right clicking the plot, one can adjust several parameters (Auto Range, Zoom, Grid Lines, Axes properties etc.) of the figure and can save the image into PNG format.
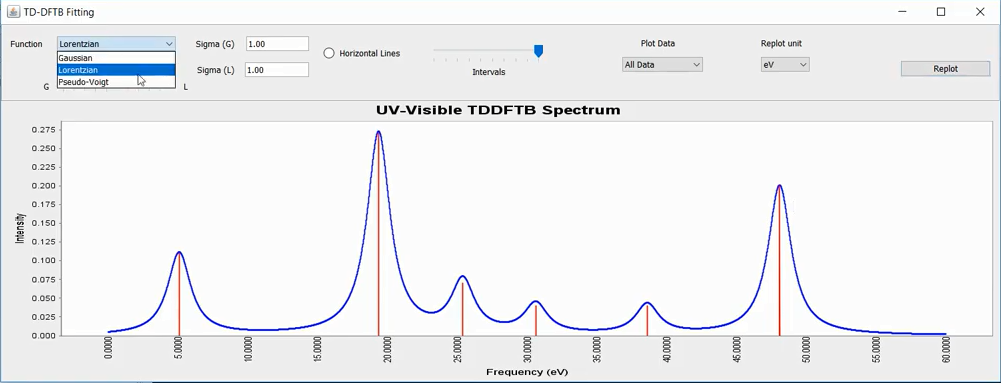
UV-Visible spectrum of an organic molecule fitted with Lorentzian convolution.¶
Partial Charges¶
DFTB+ caclulates partial charges and this charges can be projected out into the individual atoms. This Utility can be invoked by:
Tools ➤ Partial Charges
This utility read both the XYZ geometry and its corresponding detailed.out file. A color scale will also be shown for a reference. Note that this will generate a file and this file be saved for a reference (See Appendix section for the Saved Data).
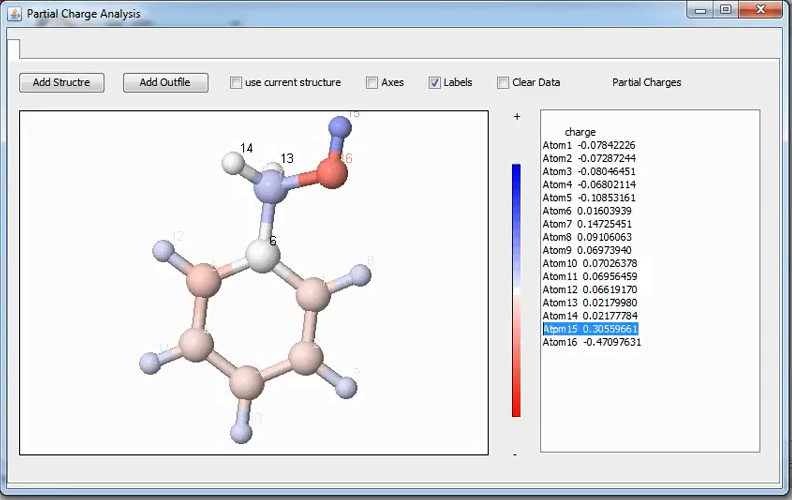
Numerical values of Mulliken’s partial charges are represented on atoms¶
Molecular-Vibrations¶
By using Harmonic approximation DFTB+ calculates the vibrational normal modes and frequencies. This requires Hessian Matrix (calculated by the DFTB+) and a script file for modes.exe binary (which calculates modes and frequencies).
The first step is to optimize the geometry. And once the optimized geometry is ready one can do the vibrational calculation with the necessary keywords (it includes Driver = SecondDerivatives{} keywords; See the example directory). This will generate the needed hessian.out file for the modes.exe.
To calculate vibrational modes vectors, to visualize/animate these modes and to get the frequencies, invoke the tool as:
Execute ➤ Run Modes
Here in this tool one has to enter the optimized GEN styled geometry
Geometry= GenFormat {
...
}
and the hessian data as:
hessian = {
...
}
Note that the dots (…) represents the numerical value of the Hessian matrix ie. ALL the numerical value of the file , hessian.out, which is 3Nx3N matrix data (force constant matrix).
After these two data entry select the corresponding SK file set and then click on the button: Generate Script. This will generate modes_in.hsd file in the ModesBinary folder. This file contains all the information for the modes.exe program which diagonalizes the hessian matrix to give modes/frequency information. The button, Generate Modes/Frequencies initiate this operation and produces the file, modes.xyz which contains the frequency/modes information.
To view this file another application is used this can be called by:
Tools ➤ View Modes
This will read the files modes.xyz and visualize the vectors/animations etc. Note that the Hessian data is used by the modes.exe binary as such. In other words it does not projects out translational and rotational motions. So that the user should be optimize the molecule well enough. If needed see http://gaussian.com/vib/ on the projection technique of Hessian matrix (Sayvetz conditions).
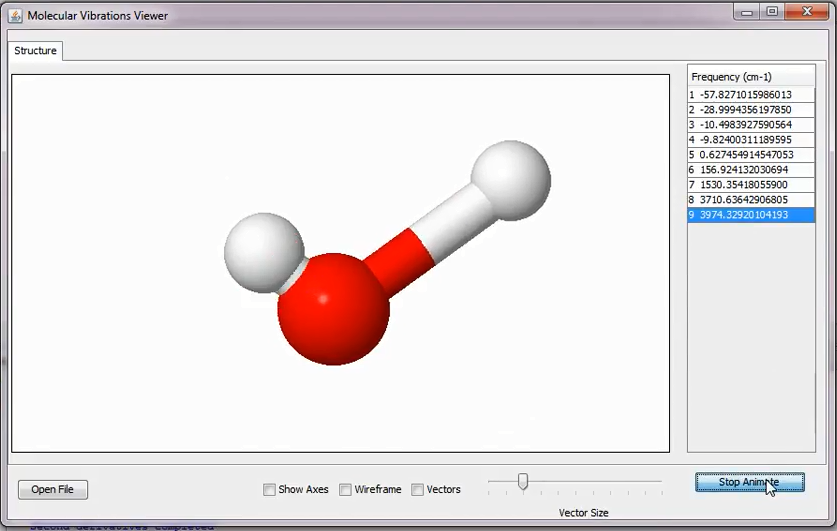
Vibrational modes of water molecules read from the file modes.xyz in the ModesBinary directory.¶
Real Time Molecular Dynamics¶
MD simulations can be done with or without using this tool. Normally one can submit MD job as usual (like a static calculation job). However, this MD tool is mainly used in real time so that a user can analyze Trajectory properties (Total Energy, Kinetic/Potential Energies, structure of each step) at Run Time. Moreover FFT based autocorrelation functions can be used to calculate IR spectra (from the dipole moments or from the velocity autocorrelation functions) after finishing the MD run. Note that, the MD analyzer tool should be invoked before the DFTB+ calculation. While running the MD calculation it will show the progress of the data along with the current structure. Its step are:
Remove all the files from ./DFTB_Scratch directory. It is Mandate.
Open MD file as, File ➤ Open (hsd)
Invoke the MD tool as, Tools ➤ MD Analyzer
Execute MD run as, Execute ➤ Run DFTB+
Wait for the MD calculation to finish; and after that use tools like, <Velocity Autocorrelation Spectra>.
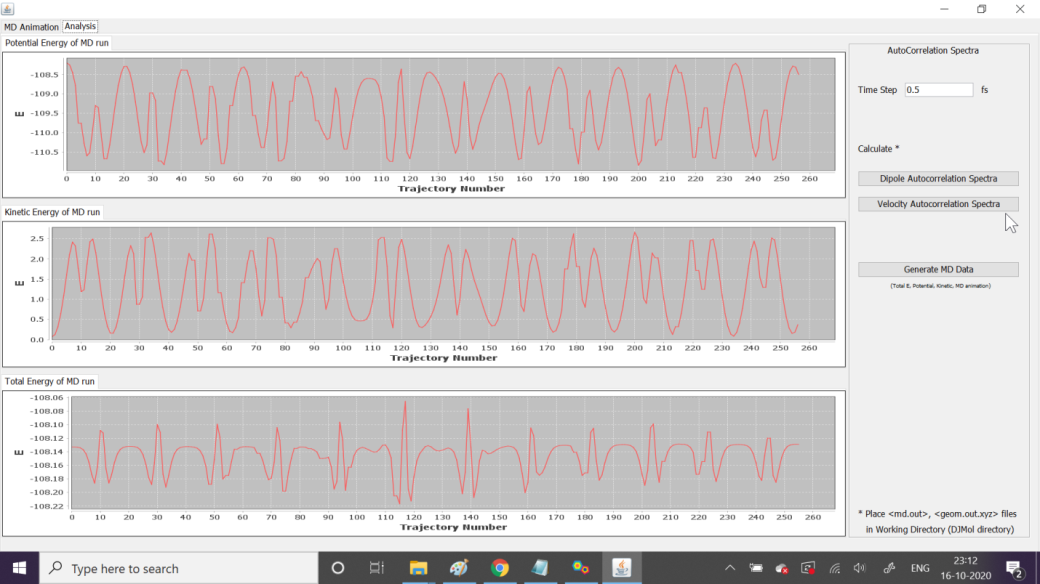
Variations of energies during the MD, in real time.¶
Potential Energy Surfaces (2D/1D)¶
The DJMol creates 2D PES (potential energy surface) by performing a series of DFTB+ calculations. For example, PES of H2O2 (hydrogen peroxide) molecule is created by doing a batch process. To create 2D PES we need two independent internal coordinate variables and, arbitrarily, we have chosen the O-O bond length and the H-H dihedral angle (in degrees) as shown in Fig. below.
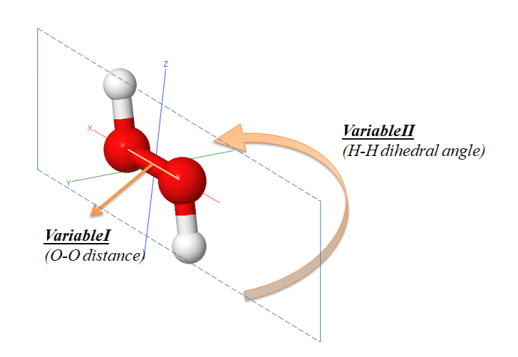
The two independent variables of H2O2 PES. The unit of bond length is in Angstrom and that of the dihedral angle is in degrees.¶
In first, make the structure of the molecule (or see Example folder for its coordinates); then using the <Convert Structure> tool (See Fig.2), convert this XYZ formatted file into the MOPIN format (which contains an equivalent z-matrix data). And copy the content of this file into <Build PES HSD> tool as shown in the Fig. 3.
By clicking the button, <Generate PES HSD Files>, this will generate 441 HSD input files for the DFTB+ program (it will take around ten minutes); copy all of these HSD files into the <SCRATCH> folder (a user can also select any other directory instead of this one) and then invoke the <Batch Processing Tool>. This will start DFTB+ program to execute all these files and create the PES in the <PESScan> folder.
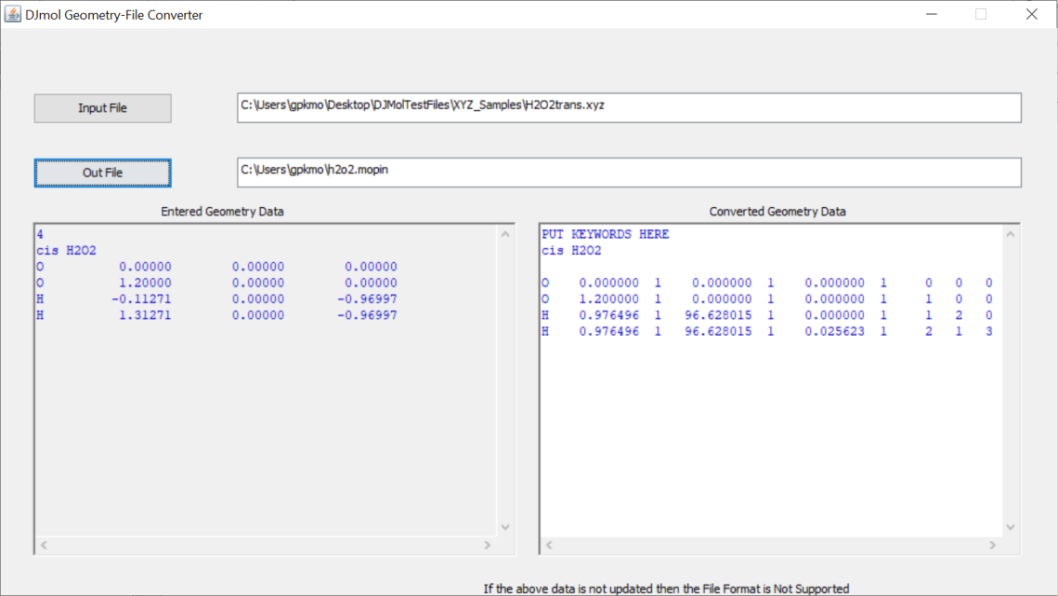
The Convert tool is used to transform Cartesian file into a Mopin file (which essentially contain the z-matrix in Mopac’s format).¶
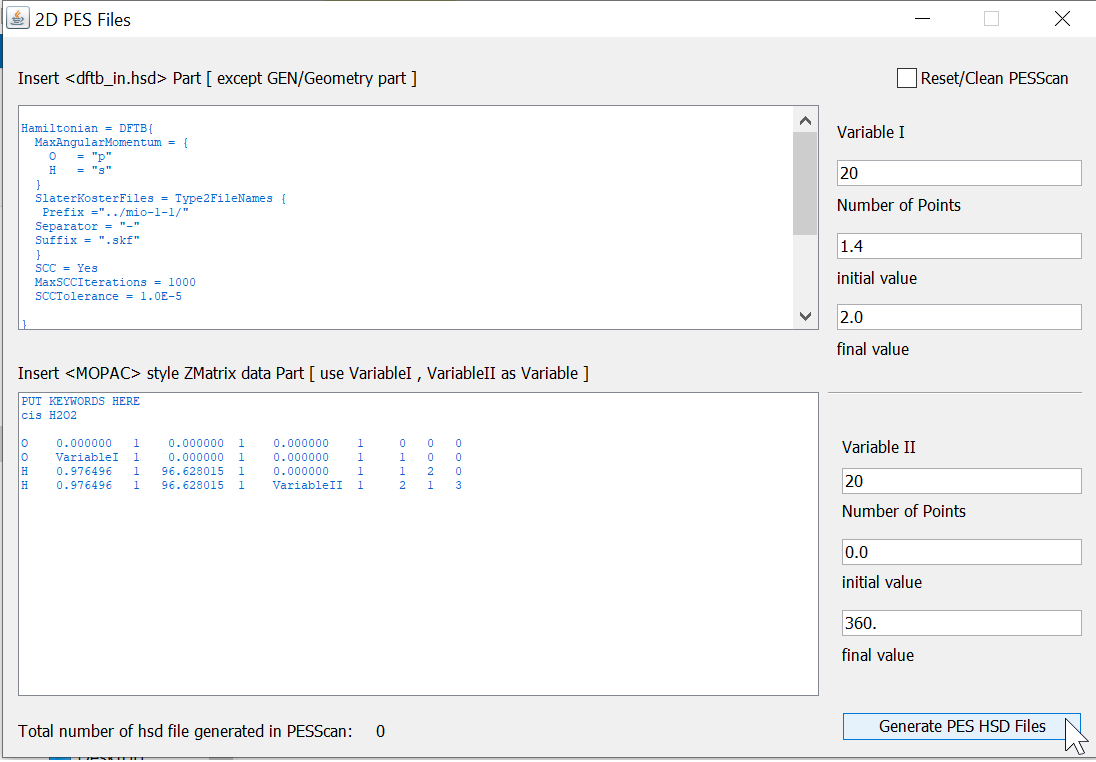
The converted MOPIN files content is inserted in the below text area and insert the variable name in the appropriate position. VariableI represents the O-O bond length (starts from 1.2Å) and VariableII, it H-H dihedral angle (starts with 0.0o).¶
After finishing the batch process, <PESViewer> can be invoked to get the plot of 2D PES or its contour diagram as shown in the Fig. 4. This utility will also indicates the minimum energy data point on the PES (in other words from this point one can start geometry optimization to locate local minimum) and its corresponding geometry file.
In <PESScan> folder one will see the following data files (See Table below).
View2DPES.dat |
Gives the data intervals in x and y directions and energy as E(x,y) in eV |
2DPESscan.dat |
It contains the sorted file names used in the PES creation with energy (in eV and in Hartree). |
2DPESscanmovie.xyz |
Contains the geometry of the individual molecules in xyz format. It can be visualized/animated with DJMol. This can be userd to construct PES with other ab initio programs. |

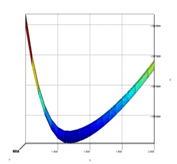
The PES of the cis-trans conversion of H 2 O 2. Note that the optimum bond length is around 1.48 Å and for shorter bond lengths the energy minimum is located near to 90.0 o or 270.0 o.¶