Chapter 3: Modeling with Siesta¶
This add-on is mainly used for performing small to moderate level Siesta DFT calculations (with hundred of atoms). For larger calculations users are advised to use cluster/GCP machines with MPI parallel binaries. This add-on is equipped with OpenMP so that multi core machines can be used for its calculations, if it is found necessary. Note that Siesta 4.1 version is used in this add-on.
At the present version all molecular structural data and non Z-matrix styled crystalline FDF data is supported by this add-on.
Siesta Script Writer¶
A basic Siesta script (.fdf file) write is written; it can be used to make a template FDF file for molecular as well as crystalline systems. Most commonly used key words are added and this can be selected optionally. However user is advised to refer Siesta 4.1 manual for finalizing the script. This tool can be invoked as:
Set Up ➤ Siesta Scripting
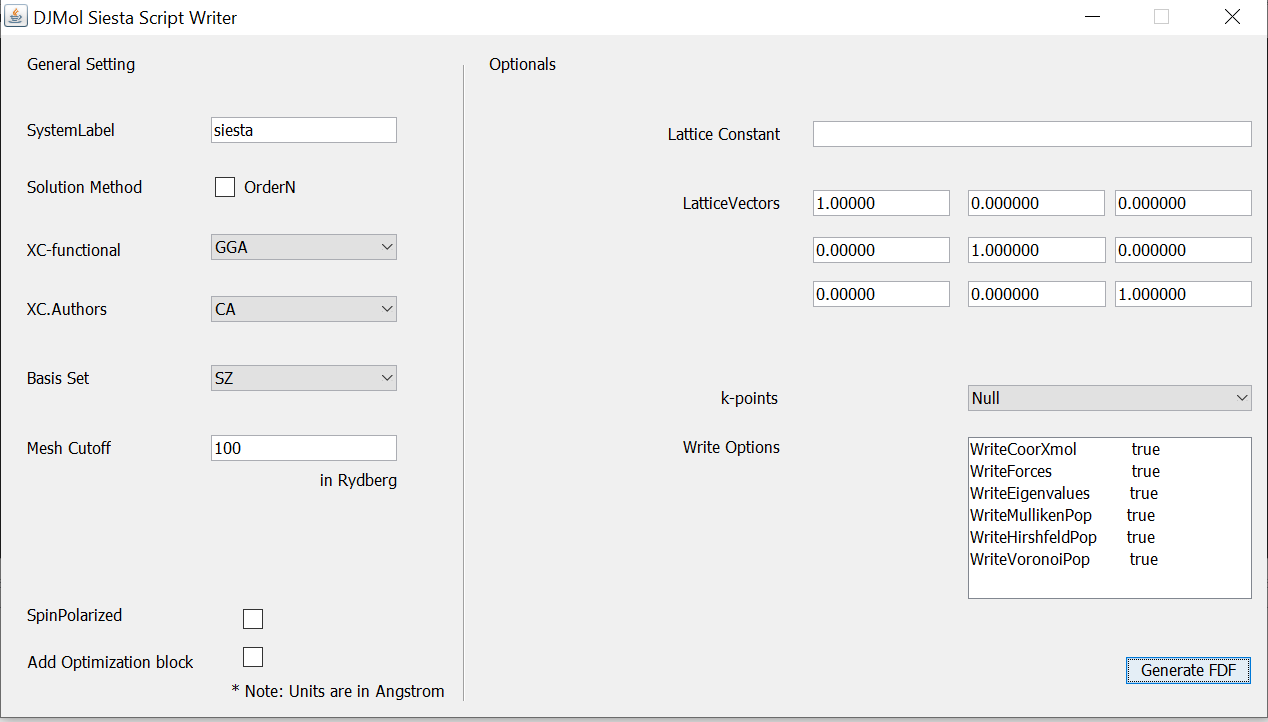
A script writer tool (FDF writer) in the Siesta add-on.¶
The newly generated file is kept in: .Input directory.
If an XYZ file is loaded into the Main Viewer, FDF file will generated with that geometry.
Siesta Add-On and Executing the Program¶
All the basic tasks for Siesta (except its script writing) is carried out in Siesta add-on and it can be called by:
Execute ➤ Siesta Calculator
It will launch the add-on as a separate program. A variety of FDF file is supported by the Jmol and hence this add-on. Use <Open> for calling these FDF files. For example, h2o2.fdf can be opened and it can be submitted in this application, by simply going to the tab <Run> and clicking the Run button. The below figure shows a console output of a Siesta run. Note that the output file is stored in, .SiestaAppSiestaBinary.4.0.CygWin64 directory. And its console log file is saved in .SiestaAppsiestaMAIN.log.
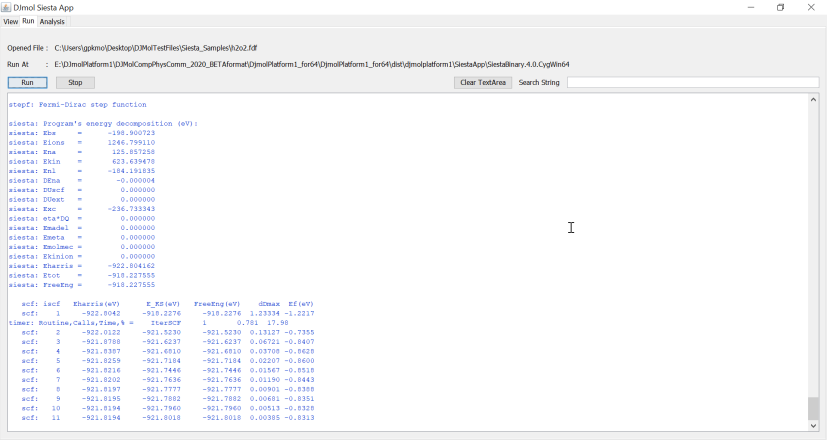
Siesta log is shown in the console window of the add-on.¶
In <Analysis> some standard or routine calculations or analysis can be done, such as checking SCF convergence, Cartesian Force components and vectors (of optimized structure), Eigenvalues etc. from the relevant files in .SiestaAppSiestaBinary.4.0.CygWin64. See the below figure of h2o2.fdf out file examples.
Basic analayzers¶
After running the calculations, one can use many basic analyzers, for example to check SCF convergence for single point as well as geometry optimization etc.
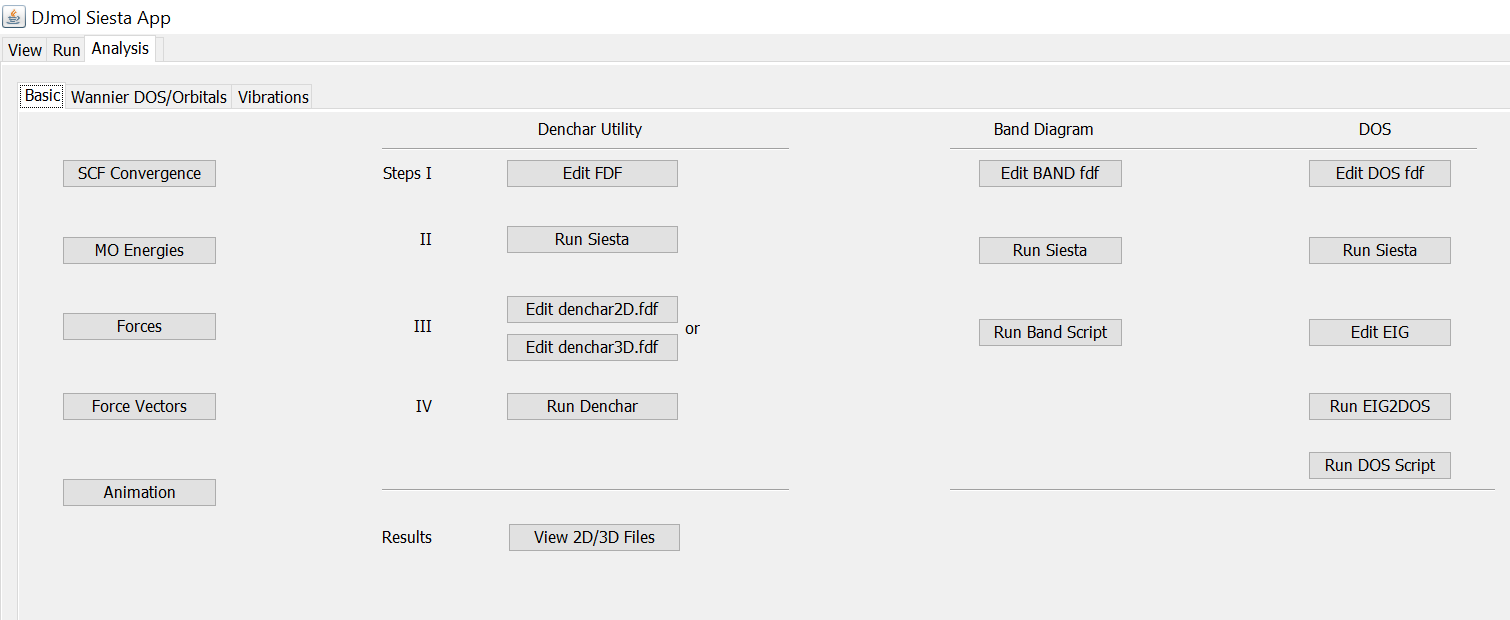
The window shows basic commands for the Siesta post processing.¶
Some of the analyzers results are shown below.
Basic Analyzers Results
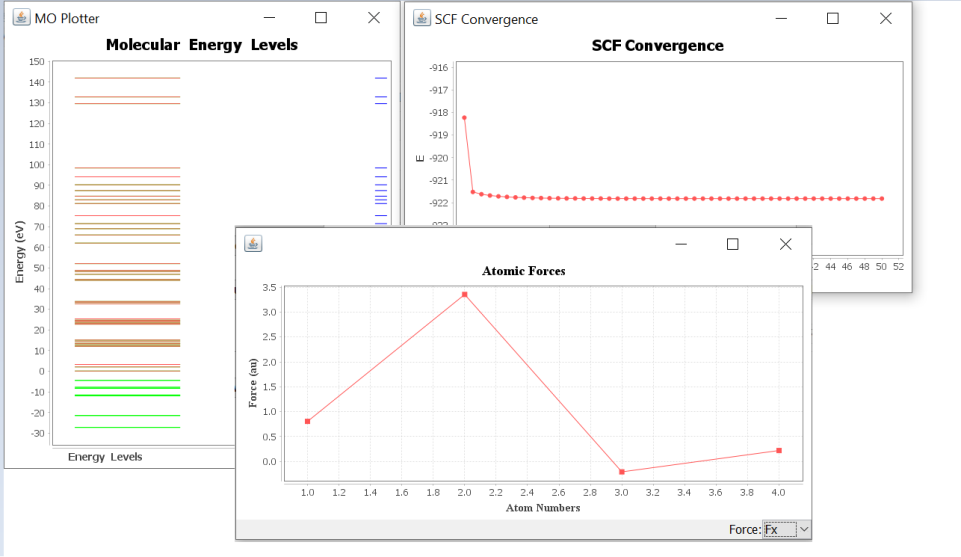
Force component vectors of each atom in H 2 O 2 (non equilibrium geometry).¶
Denchar Results
Denchar is a Siesta utility-program to plot charge densities and wave functions in real space. It can be used in 2D or in 3D. A template script for water molecule is given (but this can be modified very easily for other molecules), and its 2D/3D data can be obtained by following Steps I-IV, systematically. Note that point used in this calculations.
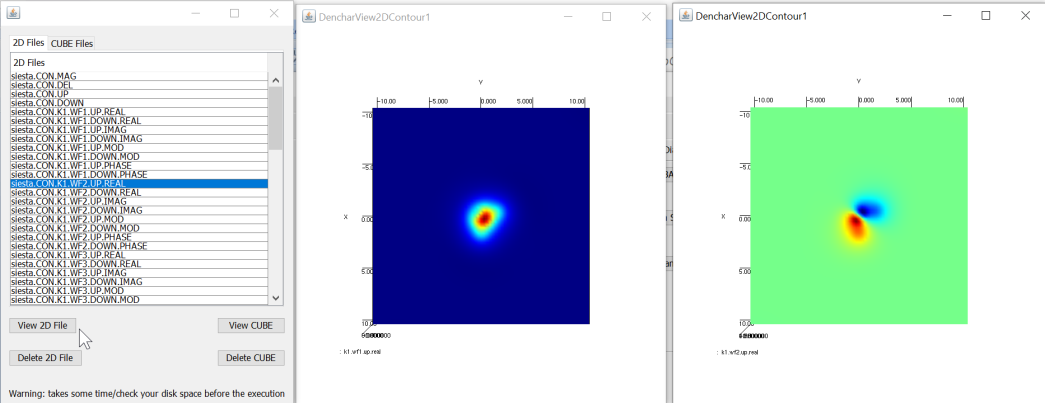
Shows list of 2D data files of water molecule from the Denchar utility, with two different real wavefunctions (the first two low lying orbitals in a contour diagram)¶
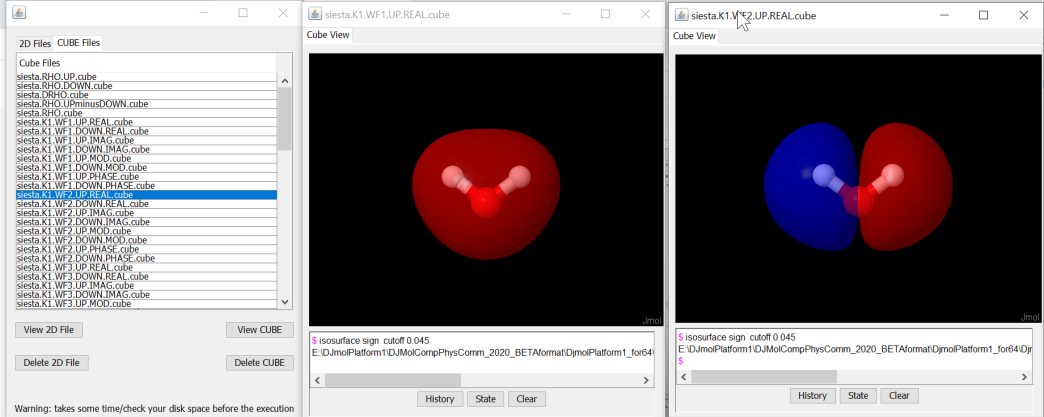
shows equivalent 3D data.¶
Similary, band diagram can also be generates with template scripts.
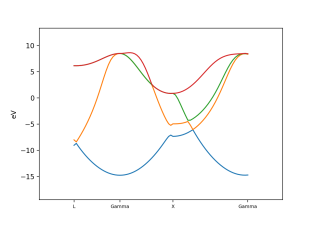
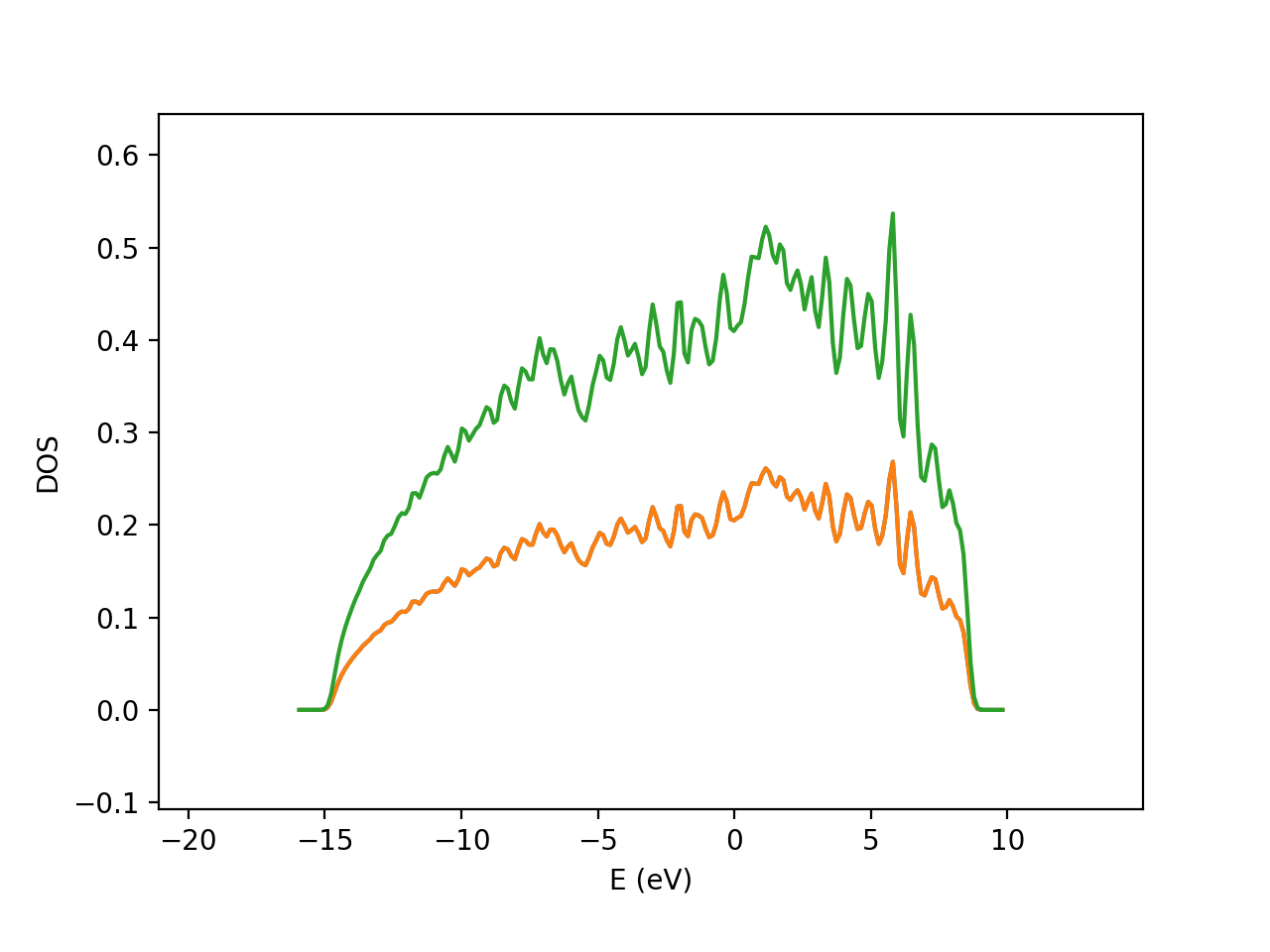
Band and DOS diagram of Aluminium (FCC). Note that in DOS plot, there are actually three lines (lower orange line is composed of two lines which corresponds to spin up and down contributions, whereas green line corresponds to the total DOS).¶
Wannier Orbitals and Band Diagrams¶
To Wannier get localized molecular orbitals of crystalline or extended systems from the Bloch wavefunction, Wannier90.exe program is used in conjunction with the Siesta binaries. See www.wannier.org for more details.
In the tab <Wannier DOS/Orbitals> this information can be obtained by following steps I to VII (as an example template, FCC bulk Si is used). Results can be viewed by using <Plot Bands> or by <View Wannier Orbitals>. This template files can be easily modified for other systems.
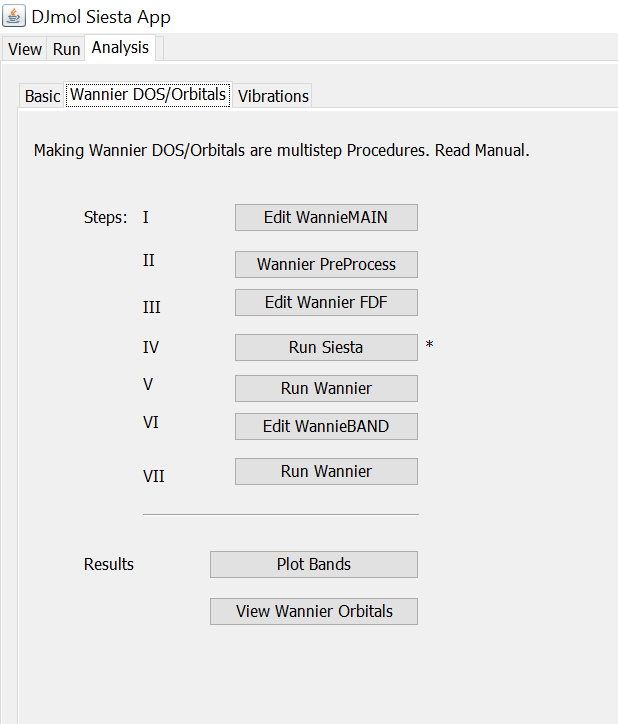
Steps of Wannier data calculations.¶
<Plot Bands> give Wannier band diagram and <View Wannier Orbitals> list XSF files and that can be converted into the CUBE file format to display Wannier functions.
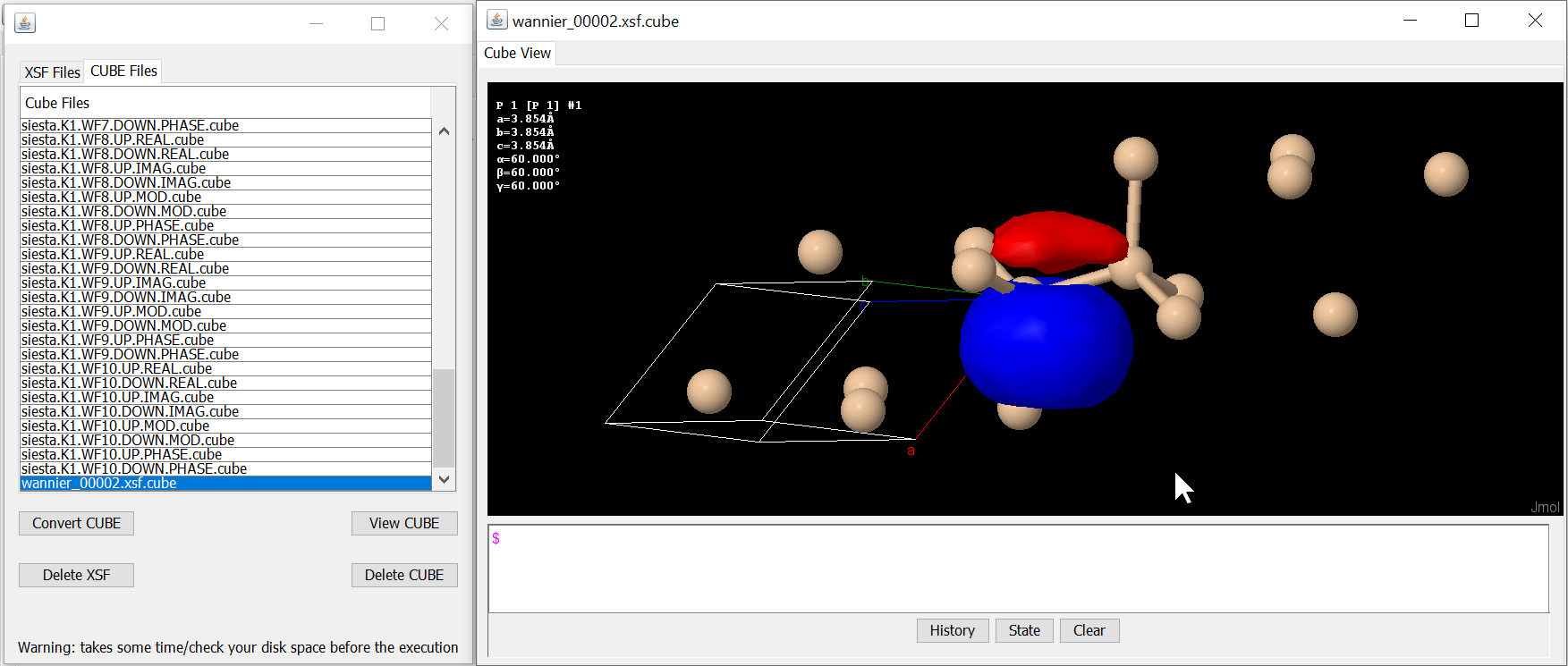
A Wannier orbital of bulk Si generated from a XSF file.¶
Molecular Vibrations¶
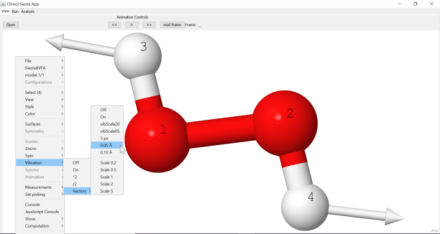
Molecular vibrations (using point) can be easily calculated with the application. In the tab, <Vibrations>, execute the steps I-V, one after another (note that example script is made for water molecule, but it can be readily modified for other molecules.). The last command, <Run Vibra> calculates eigenvalues/vectors and it gives an another window in which different modes can be animated. The essential molecules vibrational data is saved in .SiestaAppSiestaBinary.4.0.CygWin64SiestaVibModes.xyz.
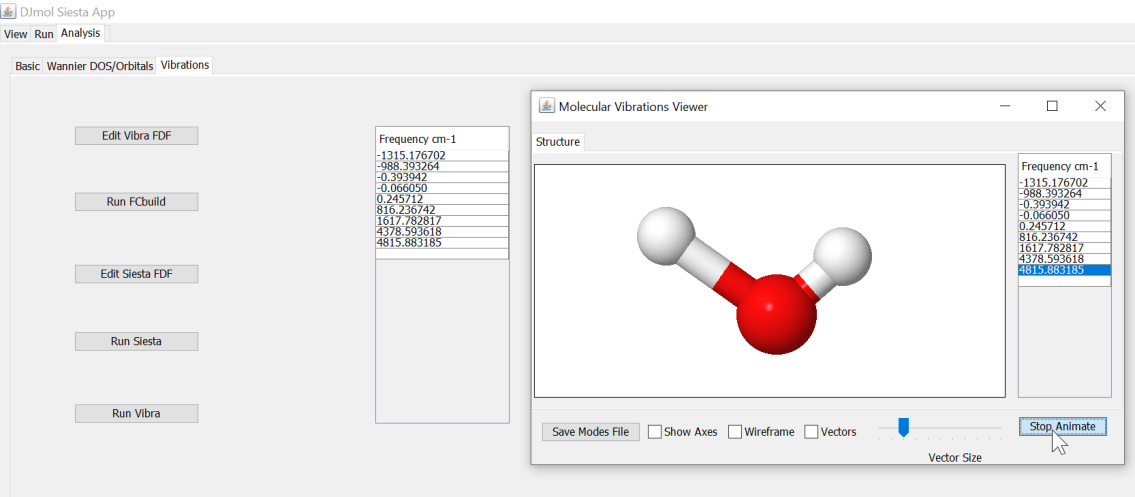
A vibrational frequency calculator module and its mode-displayer.¶