Chapter 4: OpenMD Simulations¶
OpenMD add-on is aimed to perform MD calculations with OpenMD binary/scripts. This tool has four parts, (1) Viewer, (2) Editor, (3) Terminal, and (4) Utilities.

Front end of the OpenMD add-on tool displays Ag nanoparticle obtained using OpenMD.¶
In viewer a normal XYZ file can be viewed (usually it is obtained from OMD dump file by using a converter, Dump2XYZ). All other standard Jmol view facilities are available here. Note that XYZ file will keep in the <OpenMD/Tempview> directory whereas the OMD file will be kept in the <./OpenMD> folder.
The Editor tool will open an OMD file. Since OMD file is a kind of XML file, a special editor is used to fold and un-fold many sections of this file as shown in the following file.
In Terminal-Tab, simple DOS commands can be executed. Note that one should add:
cmd.exe /c
before the Python scripts (eg. cmd.exe /c python.exe slabBuilder.py). The commands are stored in a stack-array and it can be easily called back (See Stored Commands).
In the Utility-Tab several tools are added (eg. <Clean OpenMD> will delete the MD out files and <Open Trajectory> will open a separate viewer window to animate trajectories etc.). The <Plot Energies> will open <Stat> file and plot total, kinetic and potential energies. Similarly, <T P V> plot variation of temperature, pressure and volume during the MD run as it is stored in the <Stat> file.
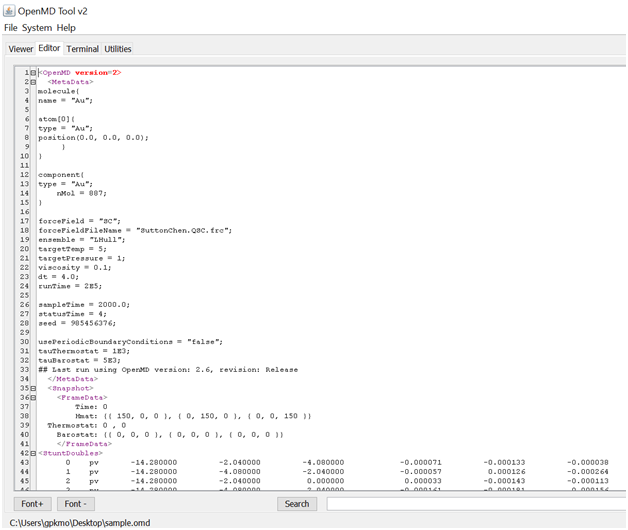
OpenMD Tool’s XML styled editor for the OMD scripts.¶
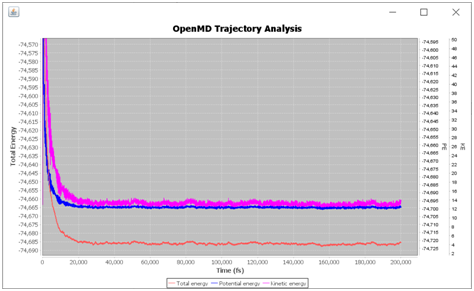
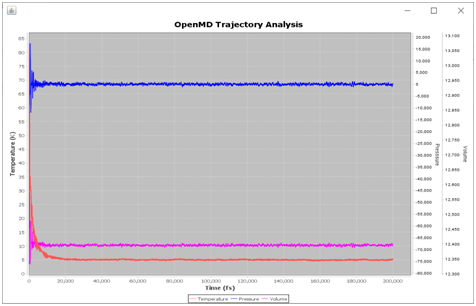
OpenMD Analyze variation of different energies during an MD simulation.¶
Since analyzing bond length gives an indication of reaction progress, <Calculate Bond Length> tool can be used to extract Bond length between two selected atoms (“Atom Number 1” and “Atom Number 2”). T his will read XYZ file which doesn’t use Periodic Boundary Conditions or PBC (as an example see, SampleAnimationTest.xyz file).
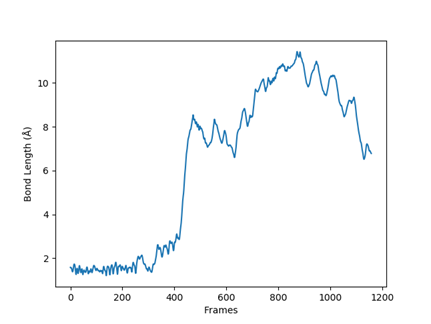
An out file from the MDAnalysis script. It describes that the bond is break near 400’th frame (in a periodic cubic box cell).¶
However, if PBC is applied more advanced script is needed to extract this bond length information. For example, if the system is equipped with Python and MDAnalysis package, a user can:
Edit <MDAnalysis> script (MDanalysisv1.py) if needed, eg. to change atom numbers
Use <Add MDAnalysis in Terminal>, which will add cmd.exe /c python.exe MDanalysisv1.py in the Terminal window
Go to Terminal and execute the above command.
The sample MDAnalysis script is:
import MDAnalysis as mda
from MDAnalysis.analysis.distances import dist
import matplotlib.pyplot as plt
import numpy as np
u = mda.Universe('mdPBCTrajectoryTest.xyz')
mybox=np.array([16., 16., 16., 90., 90., 90.], dtype='f')
# 1-2'th BL atoms in XYZ
distances = []
for ts in u.trajectory:
distances.append(dist(mda.AtomGroup([u.atoms[0]]),mda.AtomGroup([u.atoms[1]]),box=[16., 16., 16., 90, 90, 90])[2][0] )
plt.switch_backend('agg')
plt.plot(distances)
plt.xlabel('Frames')
plt.ylabel('Bond Length (Å)')
plt.show()
plt.savefig('1-2.png')
We strongly recommend to use MDAnalysis tools to post process the OpenMD data.
The details of MDAnalysis package is available at: https://www.mdanalysis.org/